- Home
- About
- Partners
- Newcastle University
- University of L'Aquila
- University of Manchester
- Alacris Teranostics GmbH
- University of Pavia
- Polygene
- Consiglio Nazionale delle Ricerche
- INSERM
- Certus Technology
- Charité Universitaet Medizin
- GATC Biotech
- University Medical Center Hamburg Eppendorf
- Evercyte GmbH
- University Hospital of Cologne
- PRIMM Srl
- University of Freiburg
- University of Antwerp
- Finovatis
- Research
- SYBIL at a glance
- Bone
- Growth plate
- Desbuquois dysplasia
- Diastrophic dysplasia
- MCDS
- Osteopetrosis
- Osteoporosis
- Osteogenesis imperfecta
- Prolidase deficiency
- PSACH and MED
- Systems biology
- SOPs
- Alcian Blue staining
- Bone measurements
- BrdU labelling
- Cell counting using ImageJ
- Chondrocyte extraction
- Cre genotyping protocol
- DMMB assay for sulphated proteoglycans
- Densitometry using ImageJ
- Double immunofluorescence
- Electron microscopy of cartilage - sample prep
- Extracting DNA for genotyping
- Grip strength measurement
- Histomorphometry on unon-decalcified bone samples
- Immunocytochemistry
- Immunofluorescence
- Immunohistochemistry
- Quantitative X-ray imaging on bones using Faxitron and ImageJ
- Skeletal preps
- TUNEL assay (Dead End Fluorimetric Kit, Promega)
- Toluidine Blue staining
- Toluidine Blue staining
- Von Kossa Gieson staining
- Wax embedding of cartilage tissue
- Contact Us
- News & Events
- Links
- Portal
TUNEL assay (Dead End Fluorimetric Kit, Promega)
Sample preparation
- sacrifice the animals and dissect knee samples
- fix in 10% neutral buffered formalin(Sigma) over 48h in the fridge
- decalcify in 20% EDTA pH7.4 for 2 weeks (shaking)
- wax embed and section (6um sections)
Note
in order to generate statistically robust data, use 3 animals per genotype, 3 sections per animal (from different anatomically matched regions in the knee), 3 sections per slide.
original protocol files:
Staining steps
dewax in xylene 2 x 5min
. ↓
100% EtOH 5min
. ↓
100% EtOH 3min
100% EtOH 3min
. ↓
90% EtOH 3min
. ↓
70% EtOH 3min
70% EtOH 3min
. ↓
50% EtOH 3min
50% EtOH 3min
. ↓
0.85% NaCl 5min
. ↓
1x PBS (rocker) 5min (from here on, do not let the sections dry!)
. ↓
4% methanol-free formaldehyde (Fisher Scientific) in PBS 15min
. ↓
1x PBS (rocker) 2x5min
. ↓
citric buffer boil (in a glass jar; 10mM citric acid, 0.05% Tween 20, pH 6.0; microwave at 800W for 4min, 400W for 4min and 200W for 4min, allow to cool down on the bench)
. ↓
1x PBS (rocker) 5min
. ↓
4% methanol-free formaldehyde in PBS 5min
. ↓
1x PBS 5min
. ↓
draw around the section with ImmEdge pen.
keep the slides in a darkened humidified chamber during all incubations
. ↓
100ul equilibration buffer 5-10min
. ↓
50ul TdT per section, 37ºC 1h
. ↓
stop the reaction in 2xSSC 15min
. ↓
1x PBS 3x5min
. ↓
mount in Vectashield with DAPI (Vector Labs)
Modifications
proteinase K unmasking in the original protocol was replaced with citric buffer boil, as proteinase K is known to generate false positives[1][2][3]
end result: nuclei showing in DAPI filter, FITC positive TUNEL positive cells
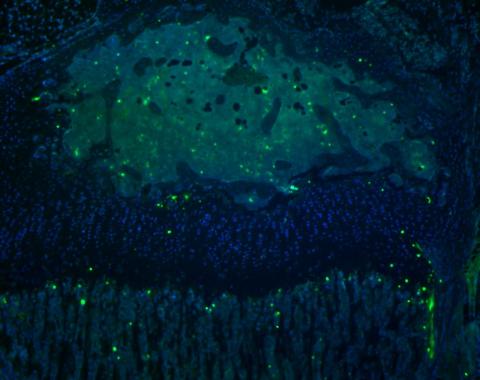
Data analysis
- count TUNEL positive cells (FITC) in the growth plate zones and present them as a percentage of all cells (FITC and DAPI) in each zone
- use One Way ANOVA to statistically analyse the data
Notes on image aquisition
Images should be taken at the right magnification, allowing identification of individual cells in each zone of the growth plate. Analysis should be perormed per channel (blue separately from green).
Procedure
Watershed algorithm can be used both in ImageJ or Fiji. Make sure java is up to date on the computer as this is a java applet.
To quantify DAPI positive cells open the file and select the zone of interest:

Clear the outline leaving only the zone of interest:

For DAPI and other fluorescence images, invert the colours:

Convert the image into an 8-bit version:

Adjust brightness and contrast (clicking “Auto” and then “Apply” is often enough):


Activate the watershed plugin:

There are certain parameters which need to be set before the analysis:

Radius indicates the predicted radius of the particles to be measured. It works well at 0.5 for the resting and proliferative zones and 1.0 for the hypertrophic zone (larger cells). Min/max level indicates the intensity of greyness which is still recognised as a particle. 0 to 200 is a good setting for the general DAPI staining. If the staining is faint and not enough cells are picked out, in particular for the hypertrophic zone, increase max to 210-220. If too many cells/particles are counted, reduce the max down to 175.
Select “8-connected” particles for the analysis. This accounts for the fact that ellipsoid shapes which will be counted can be touching but will still be included in the analysis (a function which the regular particle counter plugin in ImageJ is lacking):

Select “Overlaid dams” from the bottom menu:

Tick “Show progression messages” and press “Start Watershed”. The output file looks like this:

If too many or not enough particles were counted, adjust the Max value and/or radius.
Note
The analysis will take very long if the Max value is 255 and over. Clicking “Create animaton” slows down the process further.