- Home
- About
- Partners
- Newcastle University
- University of L'Aquila
- University of Manchester
- Alacris Teranostics GmbH
- University of Pavia
- Polygene
- Consiglio Nazionale delle Ricerche
- INSERM
- Certus Technology
- Charité Universitaet Medizin
- GATC Biotech
- University Medical Center Hamburg Eppendorf
- Evercyte GmbH
- University Hospital of Cologne
- PRIMM Srl
- University of Freiburg
- University of Antwerp
- Finovatis
- Research
- SYBIL at a glance
- Bone
- Growth plate
- Desbuquois dysplasia
- Diastrophic dysplasia
- MCDS
- Osteopetrosis
- Osteoporosis
- Osteogenesis imperfecta
- Prolidase deficiency
- PSACH and MED
- Systems biology
- SOPs
- Alcian Blue staining
- Bone measurements
- BrdU labelling
- Cell counting using ImageJ
- Chondrocyte extraction
- Cre genotyping protocol
- DMMB assay for sulphated proteoglycans
- Densitometry using ImageJ
- Double immunofluorescence
- Electron microscopy of cartilage - sample prep
- Extracting DNA for genotyping
- Grip strength measurement
- Histomorphometry on unon-decalcified bone samples
- Immunocytochemistry
- Immunofluorescence
- Immunohistochemistry
- Quantitative X-ray imaging on bones using Faxitron and ImageJ
- Skeletal preps
- TUNEL assay (Dead End Fluorimetric Kit, Promega)
- Toluidine Blue staining
- Toluidine Blue staining
- Von Kossa Gieson staining
- Wax embedding of cartilage tissue
- Contact Us
- News & Events
- Links
- Portal
COMP
Cartilage oligomeric matrix protein (COMP) is a 550 kDa large pentameric extracellular glycoprotein found in the extracellular matrix of developing and mature cartilage, within and around the tendon, in ligament and synovium and in the vitreous of the eye. The COMP gene has been located to human chromosome 19 (19q13.1). By analogy in structure it has been classified as a member of the thrombospondin protein family and as such is known as thrombospondin-5. The structure of the thrombospondin family of proteins is conserved throughout the five members. They consists of an oligomerisation domain, followed by 3-4 EGF-like domains and an invariant number of TSP type 3 sequence repeats forming 6-8 domains, the number depending on how the amino acid sequence is aligned. PSACH and MED causing mutations found to date cluser in the thirombospondin (type 3) repeat (green on the diagram below) and in the C-terminal domain (yellow) of the molecule, as seen on the diagram of COMP monomer below:
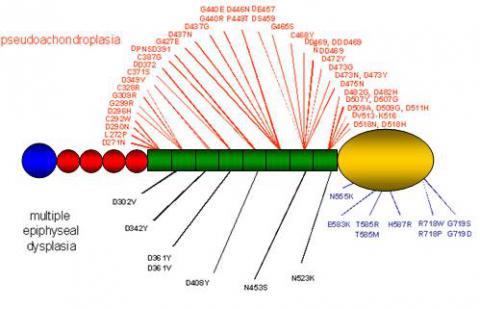
Mutations in COMP account for more than 99% of PSACH and up to 38% of the MED spectrum. Mutations found in COMP which result in PSACH and MED are predominantly located in the TSP type 3 repeats (exons 8-15, 95%). They include in-frame deletions, duplications and missense mutations. However, missense mutations resulting in MED or mild PSACH phenotypes have recently been described in the C-terminal domain of COMP and comprise 5% of the COMP mutation spectrum.
Electron microscopy of cartilage from PSACH patients with TSP type 3 mutations shows the concentration of abnormal COMP in the rER of the cells and the formation of large inclusion bodies, sometimes with a distinct lamellar structure. This ultimately causes cell death and an overall reduction of viable cells in the affected tissue. Morevoer, a disruption to the overall tissue and extracellular matrix architecture has been described, including collagen fibril orientation and morphology. This is may be due to the secondary effect being the retention of other extracellular matrix proteins normally interacting with COMP, such as type IX, XI and XII collagen, decorin, fibromodulin and others; or due to the presence of small amounts of the mutant COMP in the extracellular environment. Solid phase binding assay studies showed reduced binding of mutant COMP to type I, II and IX collagen, even though the binding site for these proteins is localised in the C-terminus. These data suggests possible interactions between the two regions and a common disease mechanism for type 3 repeats and C-terminal mutations.
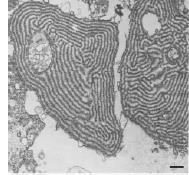 Maddox et al, J Biol Chem (1997) 272(49):3093-7
Maddox et al, J Biol Chem (1997) 272(49):3093-7
Other tissues affected by the mutations in TSP type 3 repeats are ligament and tendon. Ligaments from patients exhibit disorganised collagen fibril network, with variable diameters and disorientation. However, no protein retention was reported in those cells when cultured in vitro. In case of tendons, contradicting reports can be found, with studies showing no protein retention or retained material in the rER of the cells.
The mechanism of PSACH and MED resulting from C-terminal COMP mutations remains largely unknown, especially the fate of mutated protein and its effect on extracellular matrix. This is primarily due to the rarity of these mutations and a difficulty in obtaining patient samples (cartilage). COMP with mutations engineered in the C-terminus was shown to be secreted from different cell lines in vitro, and in an in vivo mouse model. Some conclusions regarding the pathomolecular mechanism of these mutations can be drawn from structural studies. C-terminal mutations mapped onto a 3D model of C-terminal globular domain of thrombospondin-1 appear to be present in distinct “pockets”, one of which co-localises with the postulated collagen binding site, suggesting that the C-terminal domain structure allows for three dimensional protein interactions, and that the mutations in that domain could be disrupting the interactions between important ECM components. Another pocket of mutations co-localises with a potential integrin binding site. Interestingly, patients with C-terminal COMP mutations exhibit mild myopathies. Abnomal collagen fibrills have been detecte din the mutant tendons and ligaments further supporting the role of COMP in collagen fibrillogenesis. Therefore, it has been postulated that the muscle weakness seen in PSACH/MED patients stems from disrupted interactions at the junctions between the muscle and tendon tissue.